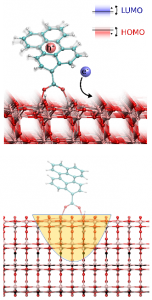
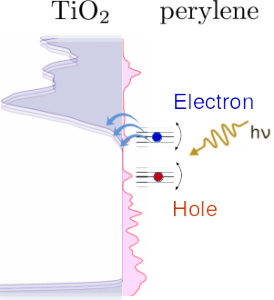
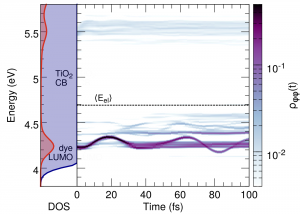
Tools for studying Dynamics of Electrons in Molecules
DynEMol: Dynamics of Electrons in Molecules DynEMol is an open-source suite for simulating nonadiabatic excited-state dynamics and molecular systems using a hybrid semiempirical approach. It has been in continuous development since 2013 and is actively maintained.
DynEMol combines two computational strategies:
Extended Hückel theory for the electronic structure
Classical force fields (AMBER/OPLS-compatible) for nuclear motion
This allows it to model a wide range of systems with nonadiabatic dynamics, including:
Molecules in vacuum or solution
Molecule–solid and solid–liquid interfaces
Bulk (crystalline and disordered) materials
- Excited-state nonadiabatic dynamics (Ehrenfest, FSSH, Ehrenfest-CSDM, and propagation in diabatic basis)
- Classical molecular dynamics
- Geometry optimization
- Force-field and extended-Hückel parameter optimization
- Time-dependent electron and hole population analysis
- Electronic and vibronic density of states (DOS, VDOS)
- Pre- and post-processing tools for simulations and analysis
Parallelization & Branches:
The repository is divided into two main branches:
-
master — optimized for HPC clusters with MPI + OpenMP support and some GPU acceleration
-
SingleNode — optimized for single-node systems, parallelized with OpenMP and supports the same GPU routines
Both branches include a folder name manipulate/ containing standalone utilities for:
- Editing input structures (copy, rotate, translate, delete atoms)
- Pre- and post-processing tools for simulations and analysis
- Generating plots from simulation output (/manipulate/general.util)
DynEMol supports a range of input formats:
-
Coordinates: .pdb
-
Force fields: AMBER/OPLS-compatible .psf, .prm, .itp, .top
-
Configuration files: card.inpt, velocity.inpt, opt_eht_parms.inpt
Input file specifications can be found in:
card_file_formats
IO_file_formats
IO_file_MM-formats
cost_file_formats
Code & Compilation:
DynEMol is primarily written in Modern Fortran (2003/2008/2018) and is designed to work best with Intel compilers:
-
Use make-ifx for Intel oneAPI (LLVM-based compilers)
-
Use make-ifort for legacy Intel compilers (pre-2022)
Other compilation targets (e.g., for debugging or profiling) are documented in the makefile.
To compile:
cp make-ifx makefile # or make-ifort, depending on your compiler
makeTo run this project, you will need to add the following environment variables to your .env file
export DYNEMOLDIR=/path/to/dynemol
export DYNEMOLWORKDIR=$(pwd)
# Place your input files in $DYNEMOLWORKDIR
$DYNEMOLDIR/dynemolExecution Script: run-dynemol.sh
The script run-dynemol.sh is a convenience wrapper for running DynEMol simulations. It handles environment setup and execution in a consistent way.
To make it accessible from anywhere in your terminal, place the script in your $HOME/bin directory:
mkdir -p $HOME/bin
cp run-dynemol.sh $HOME/bin/
chmod +x $HOME/bin/run-dynemol.shMake sure $HOME/bin is in your PATH (add this to your .bashrc or .zshrc if it’s not already):
export PATH=$HOME/bin:$PATHOnce configured, you can run DynEMol from any project directory with:
run-dynemol.shThis will execute the simulation using the input files located in the current working directory. Outputs will be saved in folders such as dyn.trunk/, dos.trunk/, MO.trunk/, etc., and refreshed at each execution.
-
Contributors:
If you have questions or encounter issues, feel free to open an issue on this GitHub repository or contact the maintainer directly at luis.gc.rego [at] gmail.com.
- Oliboni, R. S.; Bortolini, G.; Torres, A.; Rego, L. G. C. A nonadiabatic excited state molecular mechanics/extended Hückel Ehrenfest method. J. Phys. Chem. C 2016, 120, 27688–27698.
- Torres, A.; Prado, L. R.; Bortolini, G.; Rego, L. G. C. Charge Transfer Driven Structural Relaxation in a Push–Pull Azobenzene Dye–Semiconductor Complex. J. Phys. Chem. Lett. 2018, 9, 5926–5933
- Rego, L. G. C.; Bortolini, G. Modulating the Photoisomerization Mechanism of Semiconductor-Bound Azobenzene-Functionalized Compounds. J. Phys. Chem. C 2019, 123, 5692–5698.
- D. A. Hoff and L. G. C. Rego, “Chirality-induced propagation velocity asymmetry,” Nano Letters, vol. 21, no. 19, pp. 8190–8196, 2021.